A very interesting and useful patent was issued this month that describes the use and recycle of the phase-transfer catalyst methyl tributyl ammonium chloride (MTBAC, C# = 13, q-value = 1.75).
As we teach in the 2-day course “Industrial Phase-Transfer Catalysis”, careful choice of phase-transfer catalyst, non-polar solvent and high ionic strength of the aqueous phase, can result in the formation of three liquid phases, in which a small volume middle liquid phase is formed that typically consists 30%-70% of phase-transfer catalyst that has two major benefits: (1) greatly enhanced reactivity versus a non-three liquid phase system and (2) suitable for economically advantageous recovery and recycle of the phase-transfer catalyst.
In this patent, the inventors added water to the reaction mixture during workup to expand the volume of what we think is a three liquid phase PTC system. The water in the workup will be discussed later in this review.
Prior to workup during the reaction, there was little free water in the system available for hydration to form an “omega phase” (thin film of saturated aqueous solution of the reacting nucleophile; term coined by Dr. Charles Liotta of Georgia Tech) since the source of water was 50% NaOH that can act as a desiccant for the organic phase due to the high hydrophilicity of sodium and hydroxide ions that are converted to chloride ions as the reaction progresses. The reactivity of such PTC systems are often sensitive to small changes in the water available to form the omega phase which in turn affects the ability of the quat cation to act as an effective phase-transfer catalyst.
We do not know if the reactivity of this particular system was so sensitive to small changes in the amount of water in the system, but hydration level is often be a consideration when optimizing such PTC applications. We show several examples of optimizing water levels in PTC systems in our 2-day course “Industrial Phase-Transfer Catalysis.”
The inventors discuss that one key for the recovery and recycle of the MTBAC was choosing just the right amount of water during workup. That amount of water should be enough to dissolve the NaCl byproduct but not add so much water that the MTBAC, normally soluble in water with low ionic strength, will dissolve in the aqueous phase that would make the MTBAC difficult to recover and recycle. The inventors impressively achieved 84% recovery of the MTBAC by simply separating the middle phase that was well designed for the workup.
The inventors compared various phase-transfer catalysts and they found that 75% methyl tributyl ammonium chloride in water (MTBAC, C# = 13, q-value = 1.75), greatly outperformed the following phase-transfer catalysts used in solid form: benzyl trimethyl ammonium chloride C# = 10, q-value = 3.1; tetraethyl ammonium chloride C# = 8, q-value = 2.0; and tetrapropyl ammonium bromide C# = 12, q-value = 1.33.
As we teach in the 2-day course “Industrial Phase-Transfer Catalysis,” MTBAC is often an optimal phase-transfer catalyst for reactivity and cost for PTC-hydroxide “T-Reactions” (transfer rate limited) with substrates that have a pKa in the range of 16-23 according to the Halpern pKa Guidelines for the evaluation and optimization of PTC applications. The pKa of propargyl alcohol is 13.6. According to the Halpern pKa Guidelines, quat cations with higher C# should work better for PTC reactions of substrates with pKa’s below 16. This is consistent with the order of reactivity of quat salts shown in the patent.
Toluene was used as the non-polar organic solvent. As we teach in our 2-day course “Industrial Phase-Transfer Catalysis,” aromatic and aliphatic hydrocarbons, such as toluene and decane, have been have been studied in detail as solvents to form 3-liquid phase PTC systems.
MTBAC is also one of the lowest cost per mole phase-transfer catalysts used commercially in volumes of more than 100 metric tons per year in a single process.
When your company requires optimization of choice of phase-transfer catalyst for simultaneous high reactivity, low cost, and effective catalyst separation, now contact Marc Halpern of PTC Organics to integrate highly specialized expertise in industrial phase-transfer catalysis with your commercial goals.
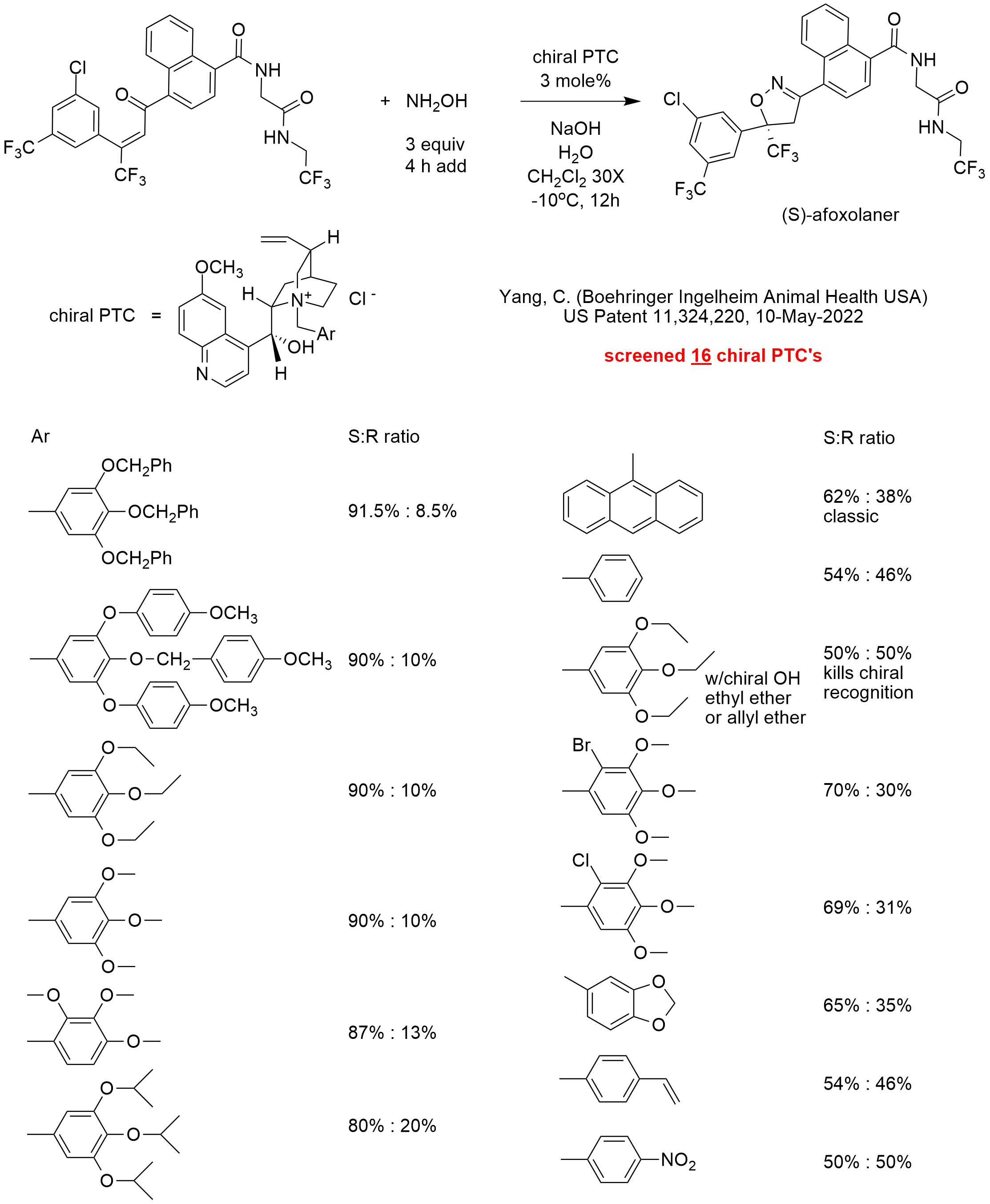
Three molecular tie points are typically required for effective chiral recognition. In many PTC chiral reactions, the three tie points include one hydrogen bond with a chiral hydroxyl and two pi-pi interactions with aromatic rings that match electron densities with or without additional steric hindrance. Every prochiral substrate is different, so each chiral PTC system must be customized for optimal selectivity.
In this patent, the inventor screened 16 chiral phase-transfer catalysts including a classic anthracenyl methyl quininium quat and a chiral ephedrinium quat (not shown in the diagram) for the addition of hydroxylamine to a double bond with a trifluoromethyl substituent.
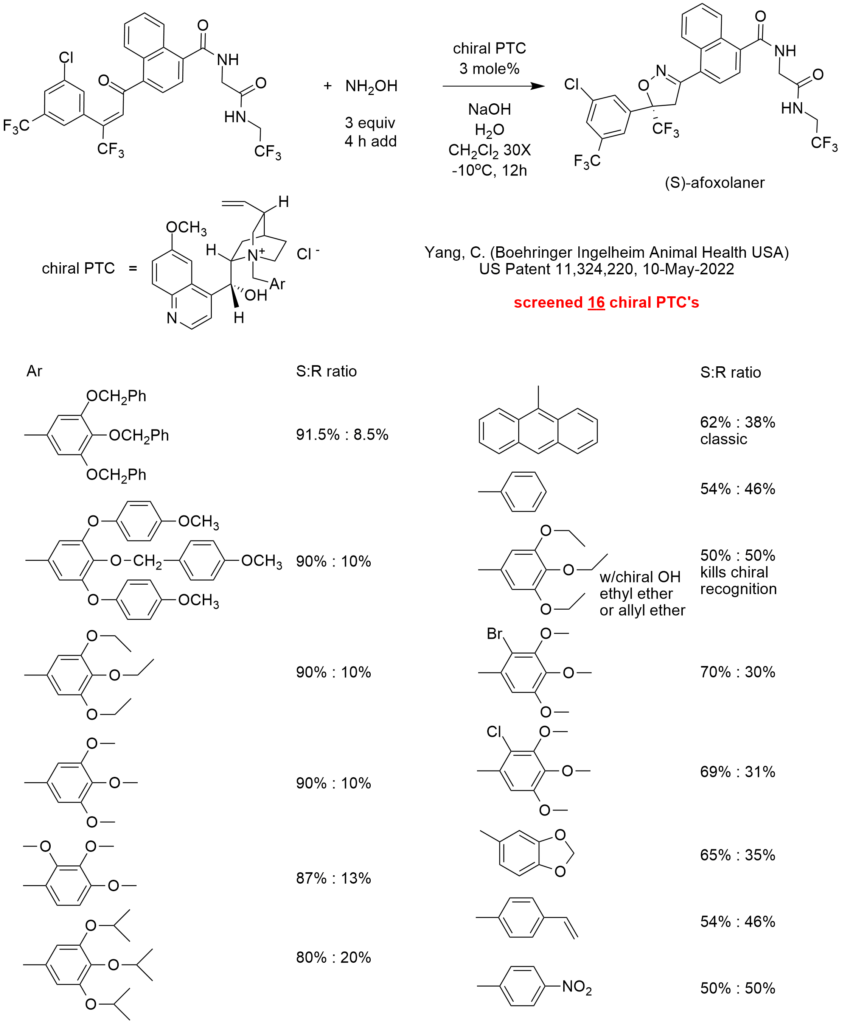
The highest performing quat was 3,4,5-tris(benzyloxy)phenylmethyl quininium chloride that gave (on a 200 g scale in Example 7) 83.6% yield of the desired (S)-afoxolaner product with > 99.0% optical purity after workup.
The diagram shows the (S) to (R) enantiomer ratio for this reaction using 16 chiral quats. The ratios are apparently for the addition reaction before or without.
Examination of the results suggest that electron-donating substituents on the benzyl group enhancements chiral recognition for this system. One may speculate that higher electron density pi-systems might increase the attraction with the electron deficient double bond with a trifluoromethyl substituent. Electron withdrawing substituents such as nitro result in total loss of enantioselectivity. Adding a bromo or chloro substituent also reduces ee. The non-substituted phenyl ring barely gives any chiral recognition (54%: 46%). When methoxy groups on the phenyl ring are replaced by isopropoxy groups, enantioselectivity decreases from 90% to 80%. This likely suggests that steric hindrance interferes with the attractive forces responsible for chiral recognition.
When the chiral hydroxy of the quinine is etherified (by allyl or ethyl), the loss of the hydrogen bonding capability totally kills chiral recognition.
Quats based on the benzylation of cinchona alkaloids often serve as effective chiral phase-transfer catalysts. Having said that, one must still customize the substituents on the benzyl ring to fine tune the electron density and steric factor to achieve optimal chiral recognition. Another example of optimizing the substitution for a chiral PTC reaction, but impressively synthesizing and screening 58 chiral diquats is described on this website at http://phasetransfercatalysis.com/ptc_reaction/highly-effective-chiral-ptc-c-alkylation-using-cinchona-alkaloid-diquats/.
Analyzing for quat salts has been an age-old challenge. Some methods work for specific situations such as colorimetry with methyl orange for tetrabutylammonium in water. Decomposition of tetrabutylammonium in the injector port of a gas chromatograph followed by detection of tributylamine has been tried but found to suffer from low reproducibility.
This month we want to highlight a newly patented method that not only analyzes a variety of quat ammonium cations, it does so using a crude sample with no pre-purification.
The invention described in Cooks; R., Jjunju; F., Li; A., Roqan; I. (Purdue Research Foundation) US Patent 11,300,555, 12-Apr-2022 provides a method for analyzing a crude oil sample by mass spectrometry in an unmodified form from which the sample was obtained. The invention provides for rapid and efficient monitoring of quaternary ammonium corrosion inhibitors in the crude oil in transmission pipelines using a bench mass spectrometer that can be deployed in the field under ambient conditions.
According to the invention a drop of crude oil containing quaternary ammonium salts is placed on filter paper and then a “discontinuous atmospheric pressure interface” (DAPI) is used with the bench-top or miniature mass spectrometer to perform the analysis.
Quaternary ammonium cations present in crude oil samples that were successfully analyzed by this method include tetrabutylammonium, tetraoctylammonium, tetrahexylammonium, tetradodecylammonium, hexadecyltrimethylammonium, benzylhexadecyldimethylammonium and alkyldimethylbenzyl ammonium.
Historically, we have not seen MS used for the analysis of quats. We assumed that this might be due to the low volatility of quat salts for applications such as GC-MS. This patent clearly demonstrates a practical system using MS to analyze quat salts in a crude non-treated oil sample.
When your company needs help in choosing a quaternary ammonium salt to achieve low-cost high-performance green chemistry using PTC, now contact Marc Halpern of PTC Organics to explore collaboration through consulting.
A reaction sequence was reported this month that uses tetrabutylammonium nitrate and tetrabutylammonium azide as reagents (not phase-transfer catalysts). Tetrabutylammonium nitrate is known to displace triflates in both primary and secondary positions. The purpose of the first step shown in the diagram is to is to invert stereocenter of the secondary carbon bearing the triflate. The second step forms the triflate again with desired stereochemistry in preparation for another inversion in the third step to produce the azide with the right stereochemistry.
The patent describes other azide displacements sometimes using sodium azide. The patent also describes the use of tetrabutylammonium nitrite (not nitrate) as reagent for deprotection of a silyloxy group.
Please note that there are 2 errors in the procedures and diagrams. The diagram states that tetrabutylammonium nitrite was used when the reagent is tetrabutylammonium nitrate based on the procedure text, the molecular weight and the known use of tetrabutylammonium nitrate for this application. Another error appears in the procedure text that cites the amount of the second triflate starting material as 41.3 mmol when it actually was 3.72 mmol. The mass of the triflate, the yield of 86% and the amount of TBA azide are consistent with 3.72 mmol and not 41.3 mmol.
While the uses of these tetrabutylammonium salts are not catalytic, one could envision using the proper choice of the right tetrabutylammonium salts with the less expensive sodium salts to reduce the cost, if cost minimization is an important factor. In such cases, you should contact Marc Halpern of PTC Organics to optimize the choice of quaternary ammonium salts as phase-transfer catalysts.
Many people confuse Aliquat 336 (trademark of BASF) and Adogen 464 (trademark of Evonik) with methyl trioctyl ammonium chloride “MTOAC.” As we explain in our article from 2012, MTOAC is one of four quat salts in Aliquat 336 and Adogen 464.
MTOAC has a molecular weight of 404 g/mole versus 432 g/mole for Aliquat 336 and Adogen 464.
MTOAC has similar properties to Aliquat 336 and Adogen 464, but since MTOAC is one quat salt and not four quat salts, it may have several advantages depending on the application.
For example, it is easier to track the fate of MTOAC, a single quat salt, than a mixture of four quat salts. This may be important for certain applications and compliance with environmental regulations.
If the reactivity of a PTC application reaches maximum activity with a PTC quat that contains 25 carbon atoms or more, then MTOAC delivers 9% more quat than Aliquat 336 or Adogen 464 for the same mass of phase-transfer catalyst due to the difference in molecular weight.
One of our affiliates in Indiana (United States) has possession of 200 kg of MTOAC more than needed. If you are interested in purchasing this MTOAC (diluted in solvent), now contact Marc Halpern of PTC Organics and he will forward your inquiry to purchase this material.
Tetrabutylphosphonium Dodecylbenzene Sulfonate was used as a “catalyst neutralizer” in the formation of polycarbonate by transesterification of diphenyl carbonate with a bisphenol and an aliphatic diol (2,2,4,4-tetramethyl-1,3-cyclobutanediol) as described in Tsunemori; H., Imazato; K., Yamanaka; K. (Teijin Limited) US Patent 11,198,759, 14-Dec-2021. The catalyst for the polymerization is a mixture of tetramethylammonium hydroxide and NaOH. The polymerization is carried out at a high temperature to distill the phenol as it is liberated during the transesterification while adjusting the pressure/vacuum in the reactor. The progress of the polycondensation is monitored and the reaction is terminated when a predetermined agitation power is reached. The catalyst neutralizer was added to the product in the molten form. We may speculate that the organophilic phosphonium quat may have been useful for dissolution of the sulfonate neutralizer. The amount of quat sulfonate added to stop the reaction is twice the molar amount of hydroxide catalyst.
As described in earlier issues of the PTC Tip of the Month, phosphonium salts are used as phase-transfer catalysts to build molecular weight of epoxy resins when reacting bisphenols with lower molecular weight epoxides (glycidyl ethers). Though not the same as building molecular weight of polycarbonate, it is interesting to note that sulfonic acids are known to deactivate phosphonium salt catalysts for epoxy resins. A reference for this is expired US Patent 4,692,504.
Now contact Marc Halpern of PTC Organics when your company can benefit from effective choice of phase-transfer catalyst or catalyst neutralizer to achieve low-cost high-performance green chemistry.
Tetrabutylammonium cyanide was used as a reagent in the two reactions shown in the diagram (not a phase-transfer catalysis reaction) to form dye compounds.
In addition to serving as a solvent, DMF can sometimes be used as a source of cyanide though in this case, tetrabutylammonium cyanide is the obvious direct source of cyanide. It appears that the cyanide adds to the unsaturated ring followed by oxidation by chloranil. If any of our readers have experience with this reaction, please share your thoughts about the mechanism of this reaction. If the role of DMF is simply to be a solvent, then we wonder if NaCN or KCN might have been sufficient as the cyanide source instead of the more expensive tetrabutylammonium cyanide.
The apparent goal of this patent is to enhance the reaction of a polyisocyanate with a polyepoxide epoxide to form a higher ratio of oxazolidinone to isocyanurate which in turn results in more desirable properties for the polymer. Since isocyanurate is formed by the self-condensation of the isocyanate, there is a competition for reaction with the isocyanate between the epoxide and the isocyanate itself.
In order to promote the reaction of the epoxide with the isocyanate, a plausible mechanism is that a halide can be used to open the epoxide ring to form a halo alkoxide anion followed by the nucleophilic alkoxide anion attacking the carbon of the isocyanate to form the oxazolidinone.
The source of the halide can be a quaternary ammonium salt. This was known previous to the patent. The novel concept disclosed in this patent is that when adding iodine to the quaternary halide, such as tetrabutylammonium bromide (TBAB) or tetrabutylammonium chloride (TBAC), the ratio of oxazolidinone to isocyanurate is higher.
The inventors compared the ratio of oxazolidinone to isocyanurate (Ox/Is) when using different ratios of TBAB or TBAC to elemental iodine. They also screened TBAB and TBAB without adding elemental iodine. The lowest (undesirable) Ox/Is ratios were observed for TBAB and TBAC without the addition of elemental iodine. The highest Ox/Is ratio was obtained with a 1:1 molar mixture of TBAB and I2.
One may speculate that tetrabutylammonium trihalide is formed such as Bu4N+ BrI2- and possibly the negative charge partially located on the iodine creates an iodide anion that is more active than a chloride or bromide due to iodide being a better nucleophile to attack the epoxide to form the iodohyrdrin alkoxide then serve as a better leaving group to enable ring closure to the oxazolidinone after the epoxide attacks the isocyanate.
If that mechanism is valid, then it would be interesting to run the reaction simply using tetrabutylammonium iodide and compare that more straightforward catalyst to the combination of TBAB/I2 or TBAC/I2. This was not reported in the patent. Since the inventors reported TBAB and TBAC without I2, one must wonder why they didn’t report TBAI without I2. If the inventors see this review, we would greatly appreciate to learn if TBAI worked poorly, unless it is not in their best interest to publicize this result.
Now contact Marc Halpern of PTC Organics to explore integrating highly specialized expertise in industrial phase-transfer catalysis with your process development program to achieve low-cost high-performance green chemistry.
Bryce Assink of W.R. Grace has been a loyal reader of the PTC Tip of the Month since about 15 years ago when he very actively participated in the 2-day PTC course in Chicago.
Bryce provided excellent insight in response to our call last month for an explanation about the identity or composition of tetrabutylammonium pyrophosphate. Shown below is Bryce’s unedited input.
Great thanks to Bryce Assink for investing his time and expertise to educate us!
————————————————————————————-
“Ok, you just made me pull out my thesis from 1999. Here’s what I have.
In 1959, Cramer and Bohm reported reaction conditions suitable for phosphorylating geraniol and farnesol to their respective diphosphates by condensation with bis(triethylammonium) phosphate, trichloroacetonitrile, and substrate alcohol in acetonitrile at rt. Reaction times were 4-24h. Diphosphate yields varied from 15-25%. (Cramer, F.; Bóhm, W. Angew Chem. 1959, 71, 775).
In 1988, Danilov modified the Cramer reaction by using Bu4NH2PO4 as a phosphate source. This reaction took 10-30 minutes, and gave ~45% yields of diphosphate for primary alcohols, and 20-30% for secondary alcohols. This was the method I used in my thesis. (Danilov, L.; Mal’tsev, S.; Shibaev, V. Soviet J. Bioorg. Chem. 1988, 14, 712).
In 1985, Poulter introduced another modification that used SN2 displacement of a tosylate or halide with tris-tetrabutylammonium pyrophosphate (ie: diphosphate) in acetonitrile. Yields were 60-90%, with no monophosphate or polyphosphate byproducts. This procedure was only useful for primary alcohols (secondary/tertiary gave elimination and/or rearrangement). (Davisson, V.; Woodside, A.; Neal, T.; Stremler, K.; Muehlbacher, M.; Poulter, C. J. Org. Chem. 1986, 51, 4768).
It looks like the patent you reference is based off of the Poulter method. My guess is that they are making the mono-quat-trihydrogen reagent themselves in the presence of 6 equivalents of water (7 after the reaction) using pyrophosphoric acid and tetrabutylammonium hydroxide. Pyrophosphoric acid is available in 94% purity. The sticky part is that this would require 73% tetrabutylammonium hydroxide (2.8 M), which does not appear to be readily available.
I hope this gives you some insight.”
A patent was issued this month that used “tetrabutylammonium pyrophosphate” as a reagent to convert the primary alcohol of derivatives of adenosine, thymidine, guanosine and cytidine into the corresponding triphosphate derivatives (such as a derivative of ATP in which the triphosphate is at the primary alcohol of adenosine) also using a chlorophosphine as a reactant that apparently provides the third phosphorous. Details of the reaction are shown in numerous examples in the patent, such as in columns 527 and 528 of Ju; J., Li; X., Chen; X., Li; Z., Kumar; S., Shi; S., Guo; C., Ren; J., Hsieh; M., Chien; M., Tao; C., Erturk; E., Kalachikov; S., Russo; J. (The Trustees of Columbia University in the City of New York) US Patent 11,085,076, 10-Aug-2021.
In order to understand the reaction and its stoichiometry, it would be valuable to understand the composition of tetrabutylammonium pyrophosphate since the pyrophosphate anion (PO3-O-PO3; sometimes called diphosphate) can have 1, 2, 3 or 4 negative charges and that means that, in principle, a quat pyrophosphate could contain 1, 2, 3 or 4 quat cations.
One of the reactions reports using 180 mg (0.33 mmol) of “tetrabutylammonium pyrophosphate” with 0.10 mmol of the adenosine derivative. That and other reactions in the patent suggest a molecular weight for tetrabutylammonium pyrophosphate of about 540-545 g/mole. However, tetrabutylammonium triihydrogen pyrophosphate would have a MW of 419 g/mole. Commercially available bis(tetrabutylammonium) dihydrogen pyrophosphate has a MW of 661 g/mole. Commercially available tris(tetrabutylammonium) hydrogen pyrophosphate has a MW of 902 g/mole. So, while we were hoping to provide insight into this reaction, we are not sure what is the composition of tetrabutylammonium pyrophosphate used in this (non-PTC) reaction.
The reaction was performed in DMF so it is possible that not much of the organophilic tetrabutylammonium cation is required to solubilize the pyrophosphate anion in the reaction phase.
We welcome any input any of our readers may have about this reaction, especially ideas about the composition of what the inventors used that they call tetrabutylammonium pyrophosphate.